Imputation Sarah Medland Boulder 2015


































52 Slides1.88 MB
Imputation Sarah Medland Boulder 2015
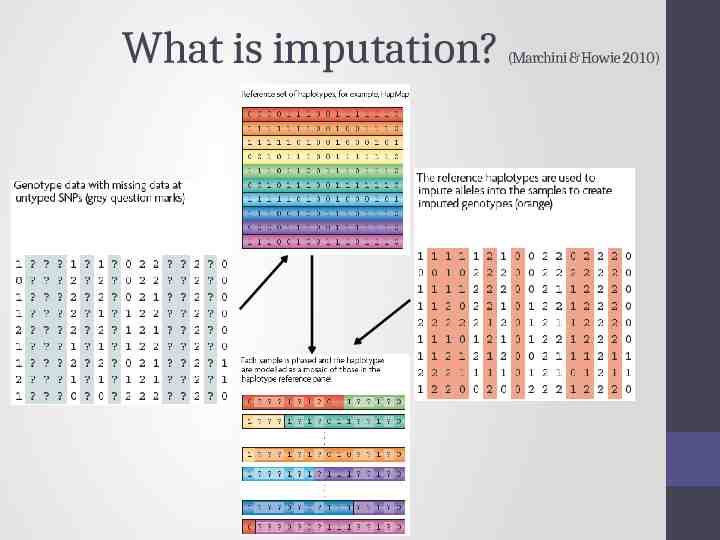
What is imputation? (Marchini & Howie 2010)
3 main reasons for imputation Meta-analysis Fine Mapping Combining data from different chips Other less common uses sporadic missing data imputation correction of genotyping errors imputation of non-SNP variation
Combining data from different chips Example 750 individuals typed on the 370K 550 individuals typed on the 610K Power MAF .2 SNP explaining 1% total variance alpha 5e-08 N 1300, NCP 13.07, power .0331 N 750, NCP 7.54 , power .0034 N 550, NCP 5.53 , power .0009
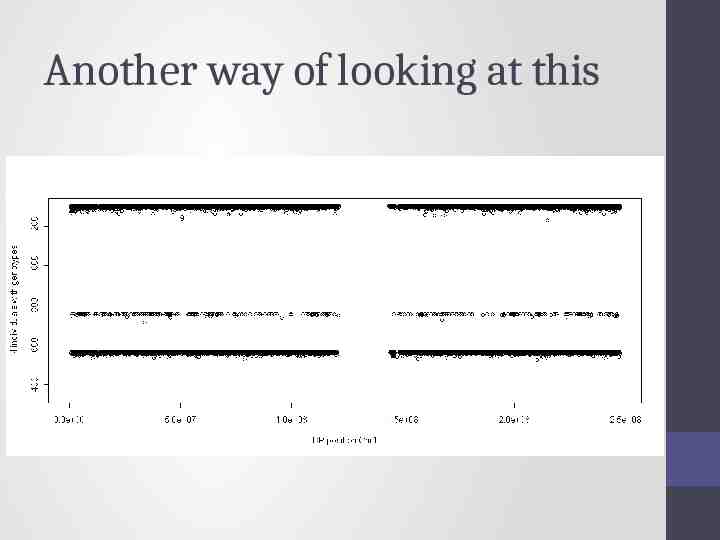
Another way of looking at this
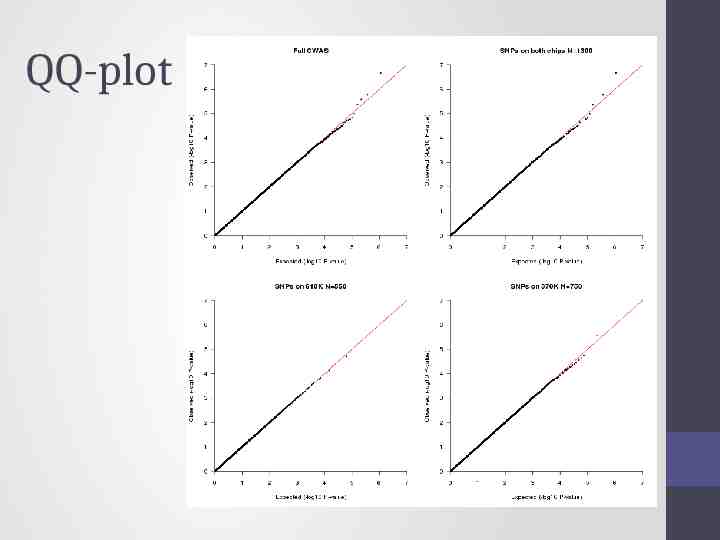
QQ-plot
Solution Impute all individuals to a single reference based on the SNPs that overlap between the chips Single distribution of NCP and power across all SNP qq plot and manhattan describes the full sample with the same degree of accuracy
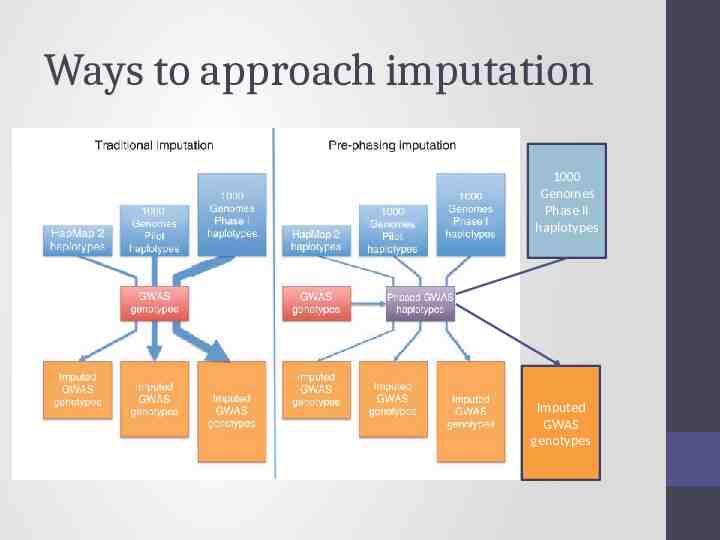
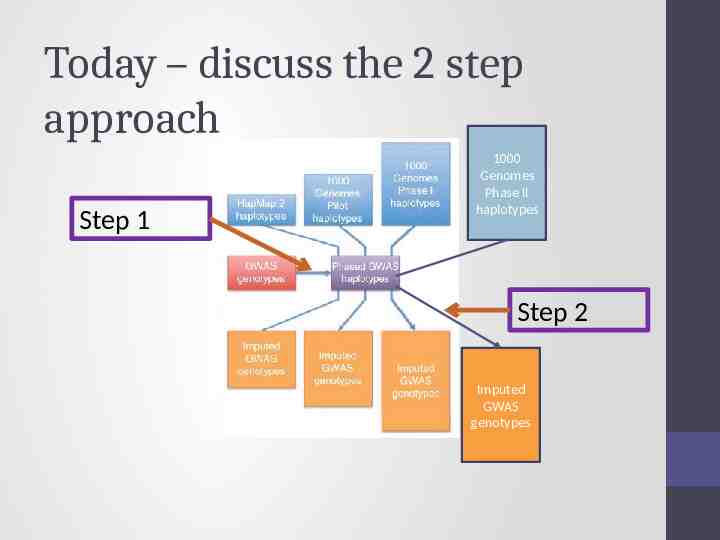
Ways to approach imputation 1000 Genomes Phase II haplotypes Imputed GWAS genotypes
Imputation programs minimac3 Impute2 Beagle – not frequently used never use plink for imputation!
How do they compare Similar accuracy Similar features Different data formats minimac3 – custom vcf format individual row snp column Impute2 – snp row individual column Different philosophies Frequentist vs Bayesian
minimac3 http://genome.sph.umich.edu/wiki/Minimac3 Built by Gonçalo Abecasis, Yun Li, Christian Fuchsberger and colleagues Analysis options raremetalworker (continuous phenotypes & family/twin samples) Format converter http://genome.sph.umich.edu/wiki/DosageConvertor Mach2qtl (continuous phenotypes) Mach2dat (binary phenotypes)
Impute2 https://mathgen.stats.ox.ac.uk/impute/impute v2.html http://genome.sph.umich.edu/wiki/IMPUTE2: 1000 Genomes Imputation Cookbook Built by Jonathan Marchini, Bryan Howie and colleges Downstream analysis options SNPtest Quicktest
Files to practice with http://genome.sph.umich.edu/wiki/Minimac3 Imputation Cookbook
Today – discuss the 2 step approach Step 1 1000 Genomes Phase II haplotypes Step 2 Imputed GWAS genotypes
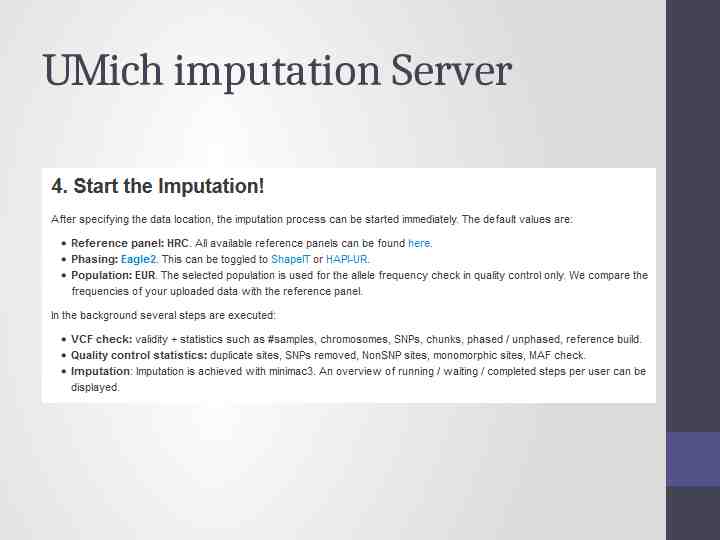
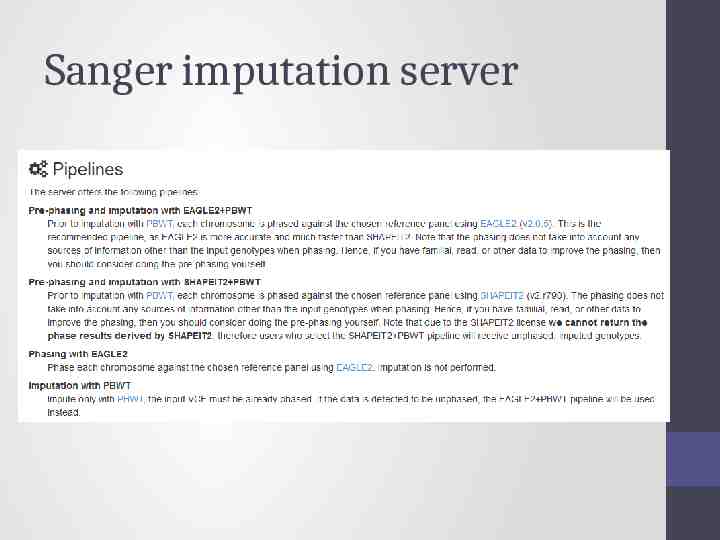
Options for imputation DIY – Use a cookbook! http:// genome.sph.umich.edu/wiki/Minimac3 Imputation Cookbook OR http://genome.sph.umich.edu/wiki/IMPUTE2: 1000 Genomes Im putation Cookbook UMich Imputation Server https://imputationserver.sph.umich.edu/ Sanger Imputation Server https://imputation.sanger.ac.uk/
UMich imputation Server
Sanger imputation server
Step 0 – Chose your reference Current Publically Available References HapMapII (no phased X data officially released) HapMapIII 1KGP – phase 1 version v3 1KGP – phase 3 version v5 Future non-public references only available via custom imputation servers HRC - 64,976 haplotypes 39,235,157 SNPs CAPPA – African American/Carabbean All ethnicities vs specific
References are in vcf format (more about this format tomorrow)
Not all references are equal!! The Beagle and IMPUTE versions of the references contain variants that do not appear in the publically available 1KGP data! The 1KGP references still contain multiple locations with more than 1 variant & Multiple variants in more than one place!
Step 0 – re-QC your data i. Convert to PLINK binary format ii. Exclude snps with excessive missingness ( 5%), low MAF ( 1%), HWE violations ( P 10-4), Mendelian errors iii. Drop all strand ambiguous (palindromic) SNPs – ie A/T or C/G snps iv. Update build and alignment (b37) v. Check strand! vi. Output your data in the expected format for the phasing program you will use Check the naming convention for the program and reference you want to use rs278405739 OR 22:395704
Strand, strand, strand DNA is a double helix A pairs with T and C pairs with G There are two strands: ATCTGGTACTCCAT TAGACCATGAGGTA Strand 1 Strand 2
Strand, strand, strand What about SNPs? ATCTGGT[A/C]CTCCAT TAGACCA[T/G]GAGGTA Strand 1 Strand 2
Strand, strand, strand What’s the big/annoying problem? ATCTGGT[A/T]CTCCAT TAGACCA[T/A]GAGGTA Strand 1 Strand 2
Strand, strand, strand Two ambiguous SNP types A/T and G/C All others are resolved How to check? Allele frequencies [know your population] LD [if you have raw data] PLINK and METAL can re-orient strand Remember the ambiguous ones!
Before we move on to talking about phasing QUESTIONS?
What is phasing In this context it is really Haplotype Estimation We take genotype data and try to reconstruct the haplotypes Can use reference data to improve this estimation
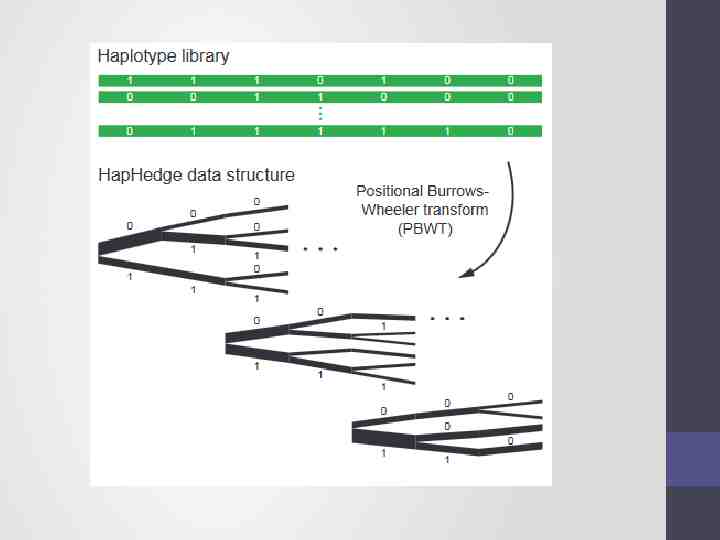
Step 1 - Phasing Input a diploid target sample and a library of reference haplotypes Selection of conditioning haplotypes. Eagle2 first identifies a subset of 10,000 conditioning haplotypes by ranking reference haplotypes according to the number of discrepancies between each reference haplotype and the homozygous genotypes of the target sample.
Generation of HapHedge data structure. Eagle2 next generates a HapHedge data structure on the selected conditioning haplotypes. The HapHedge encodes a sequence of haplotype prefix trees (i.e., binary trees on haplotype prefixes) rooted at a sequence of starting positions along the chromosome, thus enabling fast lookup of haplotype frequencies
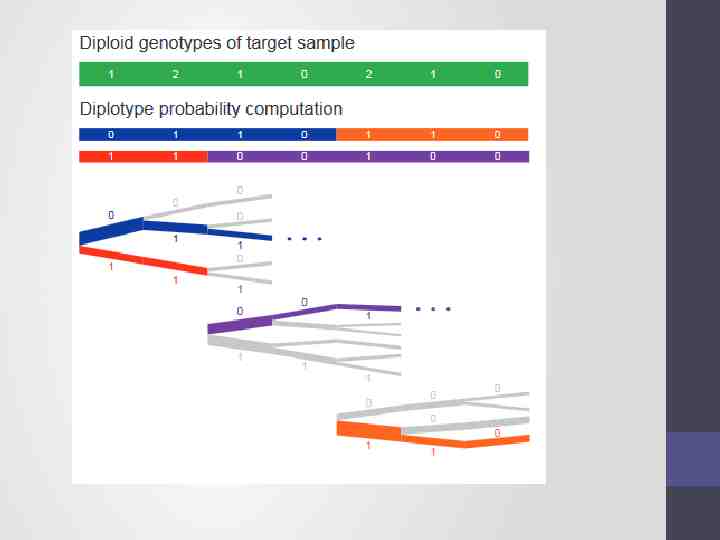
Exploration of the diplotype space. Having prepared a HapHedge of conditioning haplotypes, Eagle2 performs phasing using a HMM Consolidates reference haplotypes sharing common prefixes reducing computation.
Step 1 – How to phase Data is usually broken into manageable chunks 20Mb Each phased independently ./eagle --vcfRef HRC.r1-1.GRCh37.chr20.shapeit3.mac5.aa.genotypes.bcf --vcfTarget chunk 20 0000000001 0020000000.vcf.gz --geneticMapFile genetic map chr20 combined b37.txt --outPrefix chunk 20 0000000001 0020000000.phased --bpStart 1 --bpEnd 25000000 --chrom 20 --allowRefAltSwap
Step 2 - Imputation Compares the phased data to the references and infers the missing genotypes. Calculate accuracy metrics ./Minimac3 --refHaps HRC.r1-1.GRCh37.chr1.shapeit3.mac5.aa.genotypes.m3vcf.gz --haps chunk 1 0000000001 0020000000.phased.vcf --start 1 --end 20000000 --window 500000 --prefix chunk 1 0000000001 0020000000 --chr 20 --noPhoneHome --format GT,DS,GP --allTypedSites
Imputing in minimac
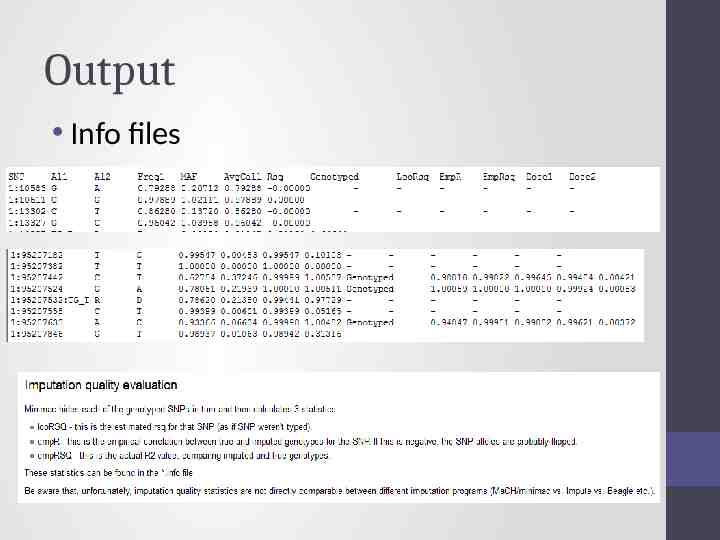
Output Info files
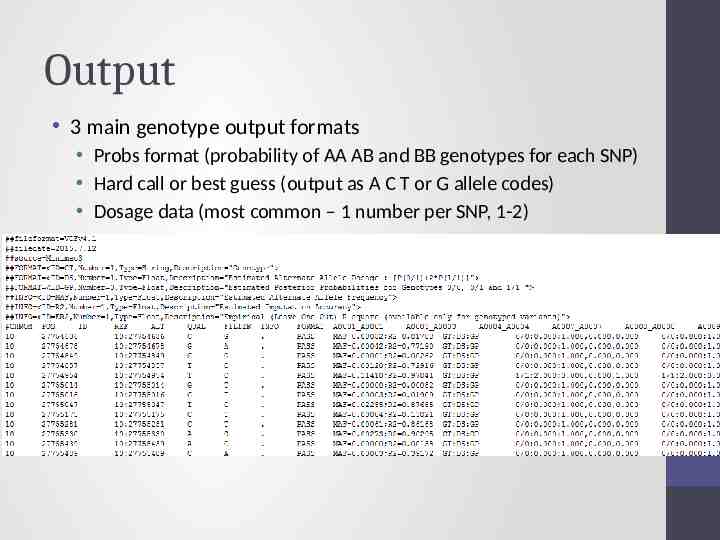
Output 3 main genotype output formats Probs format (probability of AA AB and BB genotypes for each SNP) Hard call or best guess (output as A C T or G allele codes) Dosage data (most common – 1 number per SNP, 1-2)
Assessing accuracy of phasing All phasing methods will make errors in the estimation of haplotypes - probability of error increases with length of imputed region Problem – some programs are designed to run on small segments others on whole chromosomes EAGLE2 currently considered the best More work needed that compares like with like
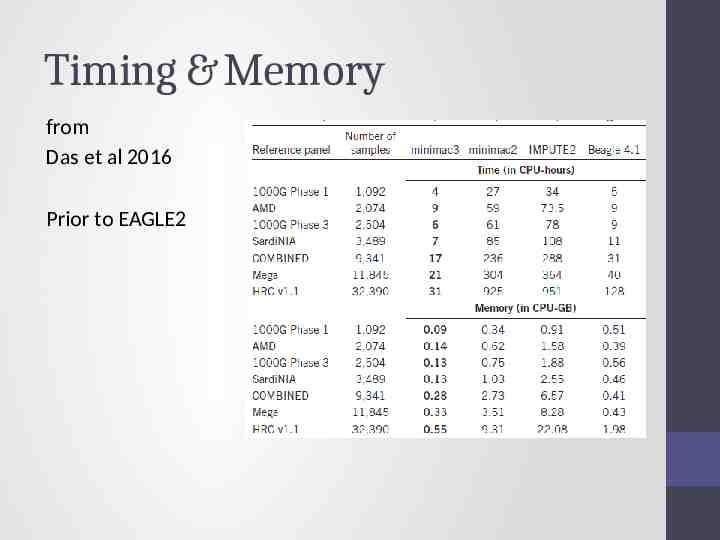
Timing & Memory from Das et al 2016 Prior to EAGLE2
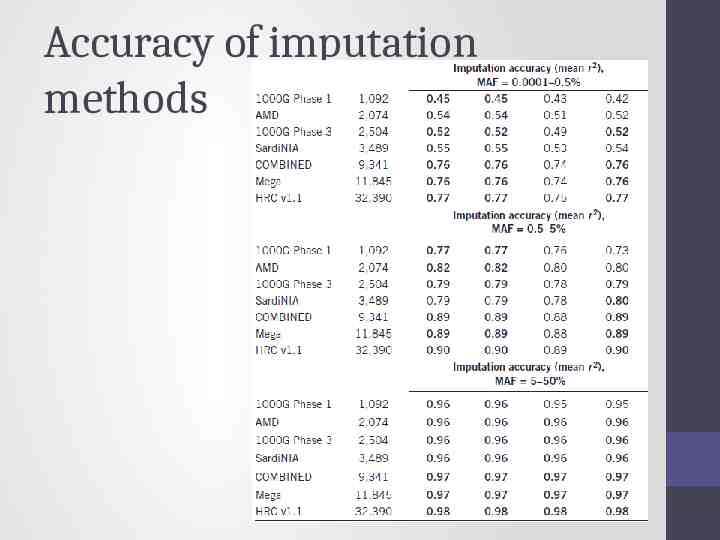
Accuracy of imputation methods
Before we move on to talking about post imputation QC QUESTIONS?
Post imputation QC After imputation you need to check that it worked and the data look ok Things to check Plot r2 across each chromosome look to see where it drops off Plot MAF-reference MAF
Post imputation QC See meta-analysis section For each chromosome check N and % of SNPs: MAF .5% With r2 0-.3, .3-.6,.6-1 If you have hard calls or probs data HWE P 10E-6 If you have families convert to hard calls and check for Mendelian errors (should be .2%) % should be roughly constant across chromosomes
Post imputation QC See meta-analysis session Next run GWAS for a trait – ideally continuous, calculate lambda and plot: QQ Manhattan SE vs N P vs Z Run the same trait on the observed genotypes – plot imputed vs observed
However, if you are running analyses for a consortium they will probably ask you to analyse all variants regardless of whether they pass QC or not (If you are setting up a meta-analysis consider allowing cohorts to ignore variants with MAF .5% and low r2 – it will save you a lot of time)
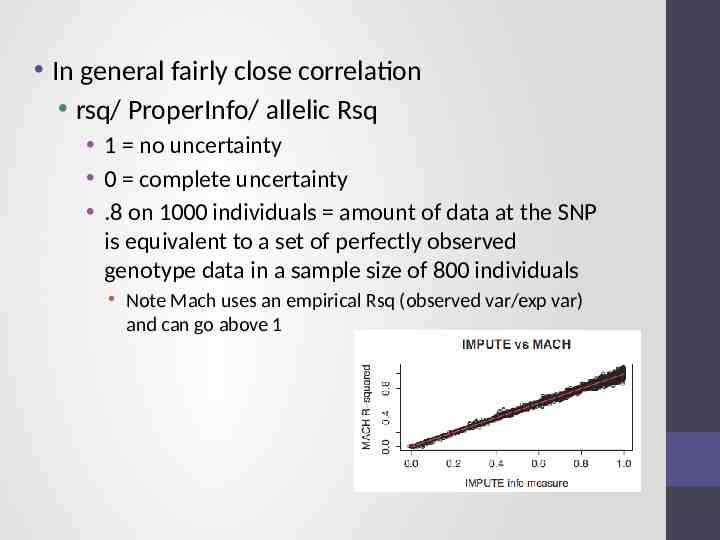
Issue – the r metrics differ between imputation programs 2
In general fairly close correlation rsq/ ProperInfo/ allelic Rsq 1 no uncertainty 0 complete uncertainty .8 on 1000 individuals amount of data at the SNP is equivalent to a set of perfectly observed genotype data in a sample size of 800 individuals Note Mach uses an empirical Rsq (observed var/exp var) and can go above 1
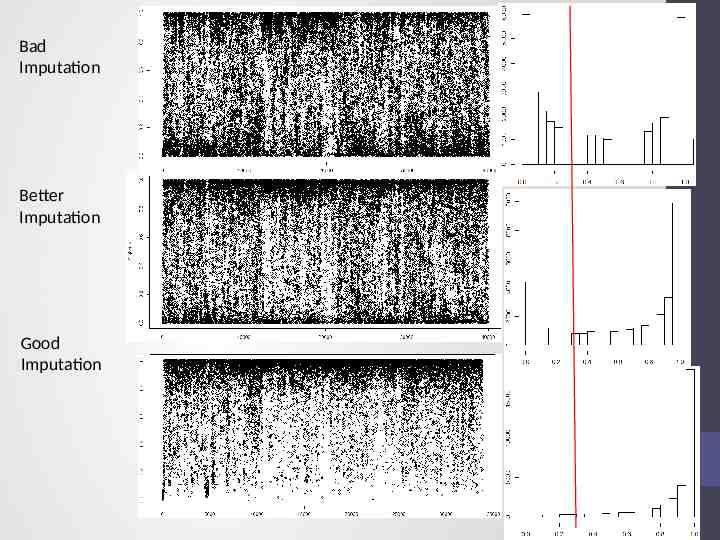
Bad Imputation Better Imputation Good Imputation
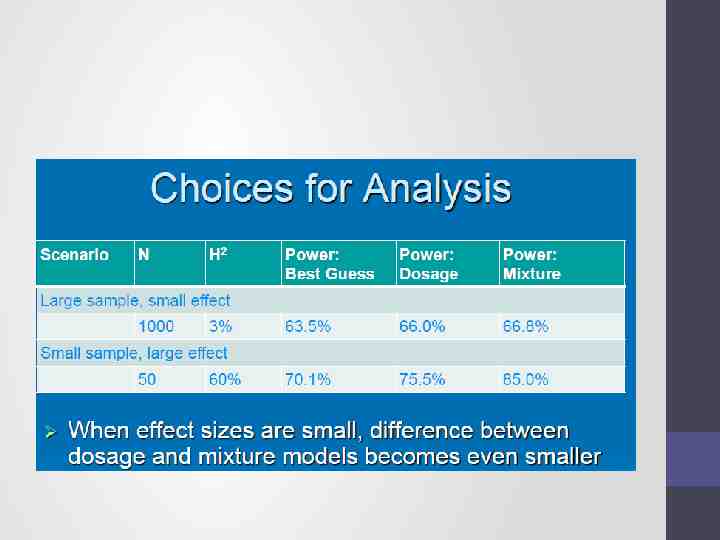
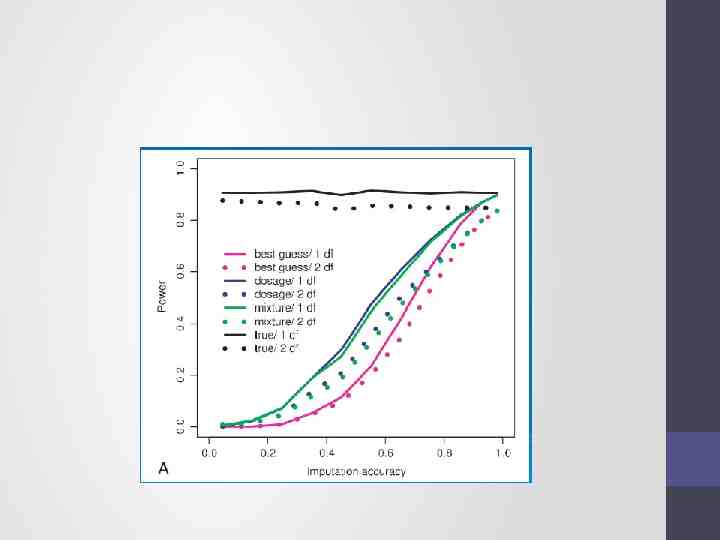
Choices of analysis methods






