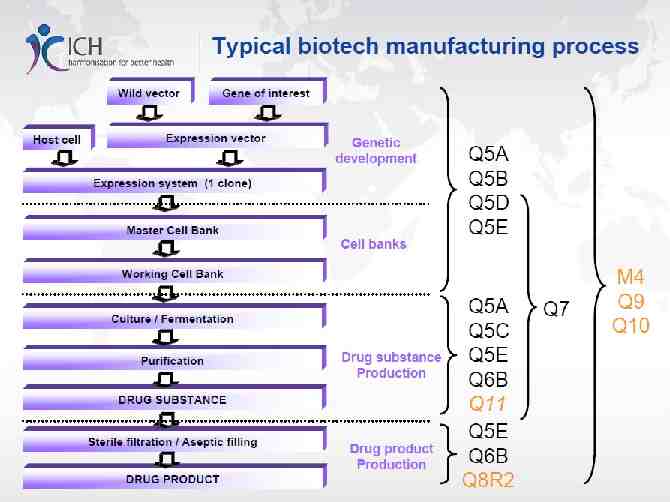
Manufacturing process of biological products: drug substance










































54 Slides2.91 MB
Manufacturing process of biological products: drug substance 9 March 2016 Sit Thirapakpoomanunt
Outline 1. 2. 3. 4. 5. 6. 7. Objective Development and Manufacturing of BP Drug substance and Drug Product GMP for Biological Products Manufacturing Process and Design Drug substance Production Technology Summary
Reference คู่มอ ื /หลักเกณฑ์การขึ้นทะเบียนตำรับยาชีววัตถุสำหรับมนุษย์(Biological Products) แบบ Asean Harmonization และ website สำนักงานคณะกรรมการ อย. WHO GMP for Biological Products Proposed replacement of: TRS 822, Annex 1(ECBS: Oct 2015,12-16 ) ICH Quality Guideline: Q7( GMP), Q8(Pharmaceutical Development), Q9(Quality Risk), Q10( Pharmaceutical Quality System) , Q11 Development and Manufacturing of DS and M4Q( Common Technical Document, Quality) Vaccine Process Technology, Biotechnology and Bioengineering, vol109,No.6, June 2012 Quality by design approach: Regulatory need, Arabian Journal of Chemistry, accepted 30 Jan 2014 Paradigm shift for vaccine manufacturing facilities: The next generation of flexible, modular facilities( Alain Pralong et al) Process Validation: General Principles and Practices, Guidance for Industry , Jan2011 CGMP
Objective To understand more about Development , Quality by design and Manufacturing Process: focus on Drug Substance Know how to use plan, do ,check and action as the continual improvement Update the process and quality control by risk assessment with scientific information Aiming to prepare study protocol for registration focus on DS Manufacturing Process
Biological Products Are medical products, made from a variety of natural sources (human, animal or microorganism). Active substances in biological products are often too complex to be fully characterized by utilizing physico-chemical testing methods alone and may show a marked heterogeneity from one preparation and/or batch to the next, include vaccines blood and blood products for transfusion and/or manufacturing into other products allergenic extracts, which are used for both diagnosis and treatment (for example, allergy shots) human cells and tissues used for transplantation (for example, tendons, ligaments and bone) gene therapies cellular therapies tests to screen potential blood donors for infectious agents such as HIV
Manufacturing Procedures Cover the manufacture, control and testing of biological products for human use, from starting materials and preparations, including seed lots, cell banks and intermediates, to the finished product. Manufacturing procedures within the scope of WHO Biological Products 2015 (replace WHO TRS 822(1992))include: a. growth of strains of microorganisms and eukaryotic cells; b. extraction of substances from biological tissues, including human, animal and plant tissues, and fungi; c. recombinant DNA (rDNA) techniques; d. hybridoma techniques; and e. propagation of microorganisms in embryos or animals.
DIFFERENT TYPES OF DEVELOPMENT NECESSARY TO REACH THE VACCINE LICENSING STAGE DS DP
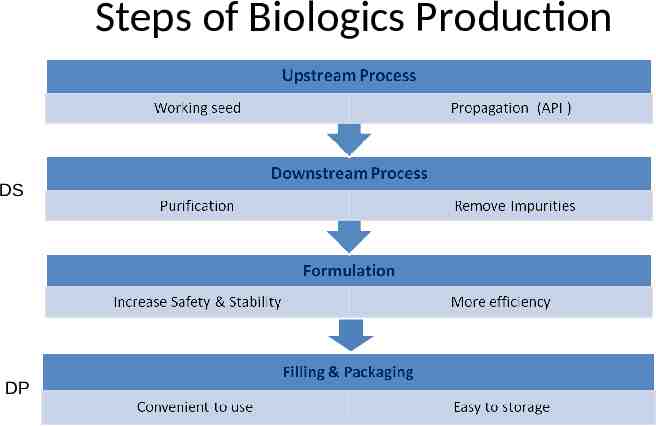
Steps of Biologics Production DS DP
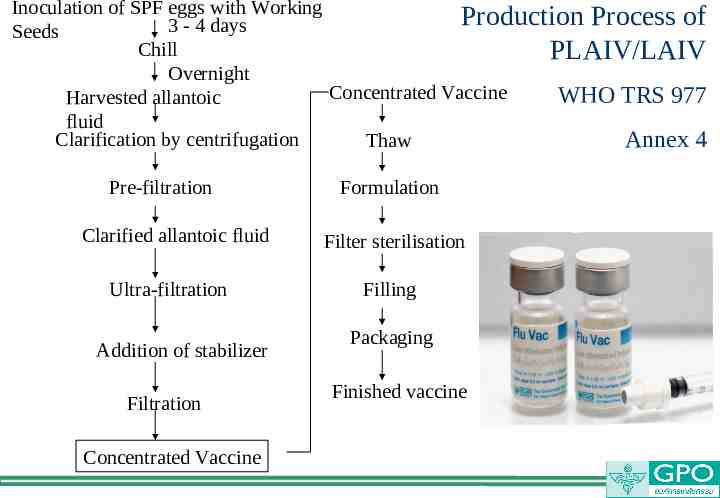
Inoculation of SPF eggs with Working Production Process of 3 - 4 days Seeds Chill PLAIV/LAIV Overnight Concentrated Vaccine WHO TRS 977 Harvested allantoic fluid Clarification by centrifugation Thaw Annex 4 Pre-filtration Clarified allantoic fluid Ultra-filtration Addition of stabilizer Filtration Concentrated Vaccine Formulation Filter sterilisation Filling Packaging Finished vaccine
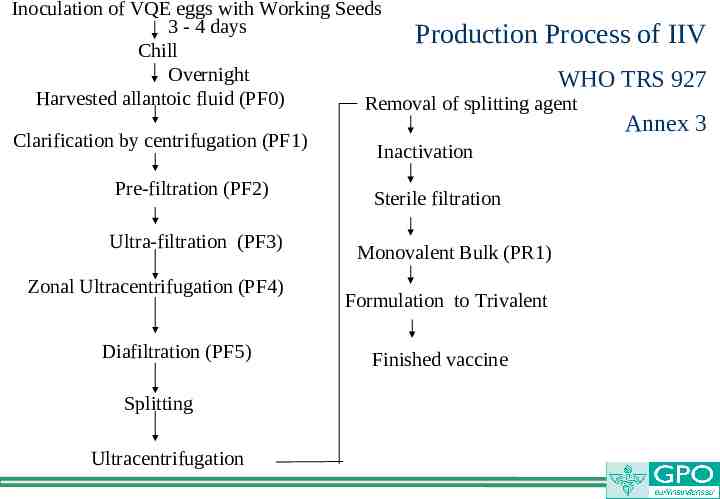
Inoculation of VQE eggs with Working Seeds 3 - 4 days Production Process of IIV Chill Overnight WHO TRS 927 Harvested allantoic fluid (PF0) Removal of splitting agent Clarification by centrifugation (PF1) Pre-filtration (PF2) Ultra-filtration (PF3) Zonal Ultracentrifugation (PF4) Diafiltration (PF5) Splitting Ultracentrifugation Annex 3 Inactivation Sterile filtration Monovalent Bulk (PR1) Formulation to Trivalent Finished vaccine
Purity, Impurities and Contaminants Purity of DS and DP is assessed by a combination of analytical procedures: due to unique biosynthetic production process and molecular characteristics DS can include several molecular entities or variants( not impurities) Impurities may be either process-related impurities(e.g. inducers,antibiotics, or media components) or Product-related impurities(e.g.precursors, certain degradation products) Contaminants include all adventitiously introduced material not intended to be part of the manufacturing process(e.g. microbial proteases) and /or microbial species. Should be strictly avoided and/or suitably controlled with appropriate in-process acceptance criteria or action limits
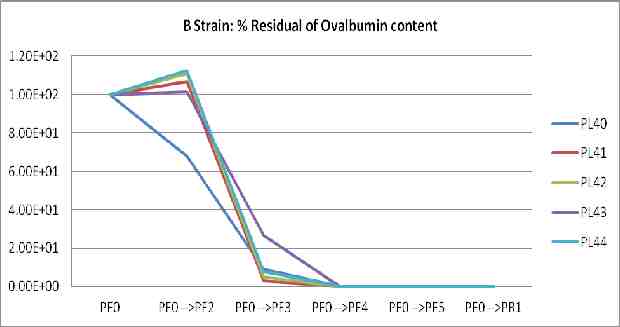
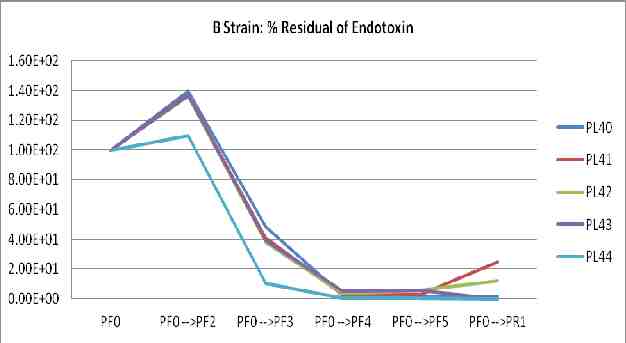
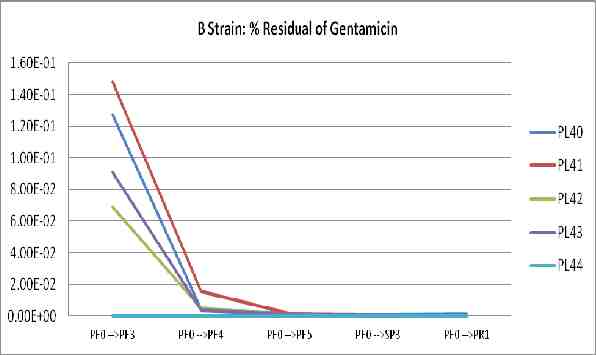
The fraction of influenza virus comparing to HA titer, endotoxin, ovalbumin, protein, and %sucrose content of Production lot IMB-H1N15809 (PL83)
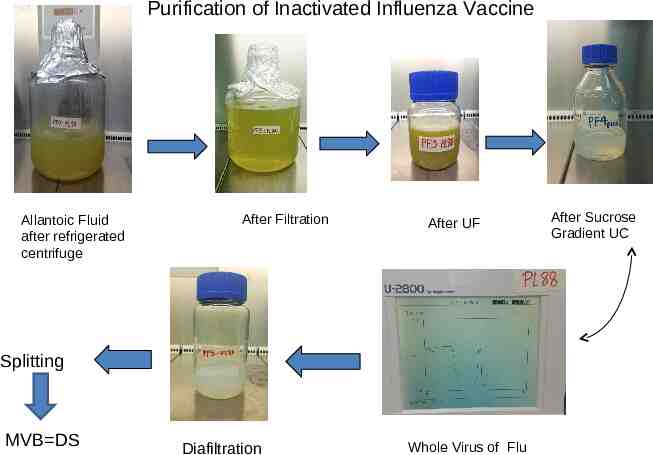
Purification of Inactivated Influenza Vaccine Allantoic Fluid after refrigerated centrifuge After Filtration After UF Splitting MVB DS Diafiltration Whole Virus of Flu After Sucrose Gradient UC
Development and Manufacturing Scope DS has been defined in ICH guidances of Q6A Specifications: Test Procedures and Acceptance Criteria(Chemical Substances) Q6B Specifications: Test Procedures and Acceptance Criteria(Biotechnological/BP) M4Q Preparation and Organization of contents of sections3.2.S.2.2-3.2.S.2.6 of Module3 of Common Technical Document
Manufacturing Process Development A. General Principles: 1. DS Quality link to Drug Product(DP) 2. Process Development Tools 3. Approaches to Development( traditional approach vs enhanced approach vs combination) 4. DS Critical Quality Attributes(CQAs) 5. Linking Material Attributes and Process Parameters to DS CQAs 6. Design Space
Manufacturing Process Development B. Submission of MPD Information 1.Overall Process Development Summary 2.DS CQAs 3.Manufacturing Process History 4.Manufacturing Development Studies
Manufacturing Process and Process Control -Description of DS manufacturing process represents the applicant’s commitment for manufacture of DS(ICH M4Q) -Should be provided in the form of a flow diagram and sequential procedural narrative -In-process control of DS for each step or stage of the process should be indicated in the description -Scaling factors should be included for manufacturing step intended to span multiple operational scales -Design space should be included as part of the manufacturing process description -Detail of batch size or scale and batch numbering should be included
Principles and General Consideration(WHO ECBS).1 The manufacture, control and administration of biological active substances and finished products require certain specific considerations and precautions arising from the nature of these products and their processes. These biological processes may display inherent variability, so that the range and nature of by-products may be variable. As a result, quality risk management (QRM) principles are particularly important for this class of materials and should be used to develop the control strategy across all stages of manufacture so as to minimise variability and to reduce the opportunity for contamination and cross-contamination.
Principles and General Consideration(WHO ECBS).2 Materials and processing conditions used in cultivation processes are designed to provide conditions for the growth of target cells and microorganisms, therefore, extraneous microbial contaminants have the opportunity to grow. Furthermore, many biological products have limited ability to withstand certain purification techniques particularly those designed to inactivate or remove adventitious viral contaminants. The design of the processes, equipment, facilities, utilities, the conditions of preparation and addition of buffers and reagents, sampling and training of the operators are key considerations to minimise such contamination events.
Principles and General Consideration(WHO ECBS).3 Specifications outlined in WHO guidelines and recommendations will determine whether and to what stage substances and materials can have a defined level of bioburden or need to be sterile. Many biological materials e.g. live attenuated bacteria and viruses, cannot be terminally sterilized by heat, gas or radiation. In addition, some products, such as certain live and adjuvanted biologicals (e.g. BCG, cholera), may not be sterilized by filtration processes. For these axenic products, processing should be conducted aseptically to minimise the introduction of contaminants from the point where a potential contamination cannot be removed from the manufacturing process.
Principles and General Consideration(WHO ECBS).4 WHO guidance documents should be consulted on the validation of specific manufacturing methods, e.g. virus removal or inactivation (20). The robust environmental controls and monitoring and, wherever feasible, in-situ cleaning and sterilization systems together with the use of closed systems can significantly reduce the risk of accidental contamination and crosscontamination.
Principles and General Consideration(WHO ECBS).5 The combination of variability in starting materials and the potential for subtle changes during the manufacturing process of biological products also requires emphasis on production consistency which becomes a special concern because of the need to link consistency to original clinical trials documenting the product's safety and efficacy. A robust manufacturing process is therefore crucial and inprocess controls take on a particular importance in the manufacture of biological active substances and medicinal products. GMP should prioritize the safety of the recipient to whom the biological product is administered, the safety of the operators during operations and the protection of the environment.
Pharmaceutical quality system(PQS) and quality risk management(QRM) The requirements of a pharmaceutical quality system (PQS) based on a life-cycle approach as defined in WHO Good manufacturing practices for pharmaceutical products: main principles This approach facilitates innovation and continual improvement, and also strengthens the link between pharmaceutical development and manufacturing activities. QRM principles should be used to develop the control strategy across all manufacturing and control stages – including materials source and storage, personnel and materials flow, manufacture and packaging, quality control, quality assurance, storage and distribution activities,
Key Components in Manufacturing Process (1) Peopleware: Persons responsible for production and control should have an adequate background in relevant scientific disciplines with sufficient practical experience Healthy with health monitoring and appropriate vaccination Training in cleaning and disinfection procedures, hygiene and microbiology Because the risks are difficult to manage, personnel working in animal facility should be restricted from entering production areas where potential risks of cross contamination exist. Staff assigned to production of bacille Calmette-Guerin (BCG) products should not work with other infectious agents. Personnel working in BCG manufacturing and animal quarters if needed to be reassigned to other manufacturing unit, should not be allowed into those areas until they pass their health check.
Key Components in Manufacturing Process (2) Software: Production technology includes .1 Starting materials(source, origin and suitability of active substances) Seed lots and cell banks - Manufacturing process, appropriate controls over the sourcing, testing, transport and storage should be in place. - Prevent the unwanted drift of genetic properties - Cell stock changes should be covered by a validation protocol and communicated to the NRA, as applicable. - An appropriately controlled environment to protect the seed lot and the cell bank and the personnel handling them. - During the establishment of the seed lot and cell bank, no other living or infectious material (e.g. virus, cell lines or microbial strains) should be handled simultaneously in the same area or by the same persons. - Each storage container should be adequately sealed, clearly labelled and kept at an appropriate temperature. - MSLs, MCBs, and preferably also WSLs and WCBs, should be stored in two or more controlled separate sites in order to minimize the risks
Software: Production technology includes(cont.).2 Production : -cultivation conditions, media and reagents are designed to promote the growth of cells or microbial organisms. - control strategy for ensuring that there are effective steps for preventing or minimizing the occurrence of unwanted bioburden, endotoxins, viruses of animal and human origin, and associated metabolites. - QRM process should be the basis for implementing the technical and organizational measures required to control risks of contamination and cross-contamination. - inoculum preparation area should be designed such as to control the risk of contamination effectively and should be equipped with a biosafety hood for primary containment. In-process Control Quality Control Validation Documents
Software: Production technology includes(cont.).3 -Growth media should be sterilized in situ by heat or in-line microbial retentive filters. Additionally, microbial retentive in-line filters should be used for routine addition of gases, media, acids or alkalis, etc., to fermentors or bioreactors. -Data from continuous monitoring of certain production processes (e.g. fermentation) should form part of the batch record. -Viral inactivation or removal process is performed, measures (e.g. related to facility layout, unidirectional flow and equipment) should be taken to avoid the risk of recontamination of treated products by non-treated products. -QRM principles should be applied to devise the control strategy regarding these pieces of equipment and associated components when used in campaign manufacture and in multi-product facilities. -Antibiotics may be used during the early stages of production to help prevent inadvertent microbial contamination or to reduce the bioburden. Acceptable residual levels should be defined
Quality Control and In-process Control: -the sampling materials being sampled in order to ensure that the testing carried out is representative. -cell-based products, microbiological tests (e.g. sterility test or purity check) should be conducted on cultures of cells or cell banks free of antibiotics and other inhibitory substances -The traceability, proper use and storage of reference standards should be ensured, defined and recorded. -All analytical methods used in the quality control and in-process control of biological products should be well characterized, validated and documented
Validation(1): -Major aspects of biological products require process and cleaning validation. -A QRM approach should be used to determine the scope and extent of validation. -All critical biological processes (e.g. inoculation, multiplication, fermentation, cell disruption, inactivation, purification, virus removal, removal of toxic and harmful additives, filtration, formulation, aseptic filling, etc.), as applicable, are subject to process validation. -Manufacturing control parameters to be validated may include specific addition sequences, mixing speeds, time and temperature controls, limits of light exposure, and containment.
Validation(2): -Critical processes for inactivation or elimination of potentially harmful microorganisms of Biosafety Risk Group 2 or above, including genetically modified ones, are subject to validation. -Process revalidation may be triggered by a process change, as part of the change control system. -Cleaning validation should be performed in order to confirm the effectiveness of cleaning procedures -The integrity and specified hold times of containers used to store intermediate products should be validated unless such intermediate products are freshly prepared and used immediately, as appropriate.
Approach to Process Validation 1.Process Design: based on Knowledge gained through development and scale-up activities 2.Process Qualification : the process design is evaluated to determine reproducibility for commercial manufacturing 3. Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control
Documentation (batch processing records): -the processing records of regular production batches should provide a complete account of the manufacturing activities of each batch of biological products. -for vaccines, a batch processing record and a summary protocol should be prepared for each batch for the purpose of lot release by the NRA. -Manufacturing batch records should be retained for at least one year after the expiry date of the batch of the biological product , longer periods may provide useful information related to AEFI and other investigations. -Starting materials may require additional documentation on source, origin, supply chain, method of manufacture and controls applied in order to ensure an appropriate level of control, including microbiological quality, if applicable. -Some product types may require specific definition of what materials constitute a batch – particularly somatic cells in the context of ATMPs(Advance Therapeutic Medicinal Products). For autologous and donor-matched situations, the manufactured product should be viewed as a batch.
Key Components in Manufacturing Process (3) Hardware: 3.1 Premises and Equipment: -When multi-product facilities involve live microorganisms and viruses, the manufacturer should demonstrate and validate effective decontamination of the previously-used live microorganisms and viruses. -Documented QRM should be carried out for every additional product in a biological manufacturing multi-product facility -Killed vaccines, antisera and other biological products – including those made by rDNA techniques, toxoids and bacterial extracts – may, after inactivation, be manufactured on the same premises provided that adequate decontamination and cleaning measures are implemented on the basis of QRM. -Validation studies should be carried out to ensure the effectiveness of cleaning, sanitization and disinfection, including elimination of residues of used agents.
3.1 Premises and Equipment: (cont.) -Logical and unidirectional flows of personnel, materials and processes, use of clean-in-place (CIP) and sterilize-in-place (SIP) systems should be considered wherever possible. -Where sterile single use systems such as bags and connectors are utilized, they should be qualified with respect to suitability, extractables, leachables and integrity. -Because of the variability of biological products and the corresponding manufacturing processes, approved starting materials that have to be measured or weighed for the production process may be kept in small stocks in the production area for a specified period of time according to defined criteria -In manufacturing facilities, the mix-up of entry and exit of personnel should be avoided through separate change rooms or through procedural controls where Biosafety Risk Group 3 and 4 organisms (19) are handled. -
3.2 Cleanrooms: -the required class/grade of clean areas for the manufacture of sterile products are defined and established -the degree of environmental control of particulate and microbial contamination of the production premises should be adapted to the intermediate or finished product -The environmental monitoring programme should be supplemented by the inclusion of methods to detect the presence of specific microorganisms used for production (e.g. recombinant yeast and toxin and polysaccharide producing bacterium). -The environmental monitoring programme may also include detection of produced organisms and adventitious agents of production organisms, especially when campaign manufacture is applied on the basis of QRM principles.
3.3 Containment(1): -Airborne dissemination of live microorganisms and viruses used for the production process, including those from personnel, should be avoided. -Adequate precautions should be taken to avoid contamination of the drainage system with dangerous effluents. -Dedicated production areas should be used for the handling of live cells capable of persistence in the manufacturing environment, for pathogenic organisms of Biosafety Risk Group 3 or 4, and/or for spore-forming organisms until the inactivation process is accomplished and verified. -Where campaign manufacture of spore-forming organisms occurs in a facility or suite of facilities, only one product should be processed at any one time. -Use of any pathogenic organism above Biosafety Risk Group 3 may be permitted by the NRA according to the biohazard classification of the organism -Production of BCG related product should take place in a dedicated area and by means of dedicated equipment and utilities (e.g. HVAC systems) in order to minimize the hazard of cross-contamination.
Containment(2): -Air-handling systems should be designed, constructed and maintained to minimize the risk of cross-contamination between different manufacturing areas, as required based on QRM principles -Primary containment equipment should be designed and initially qualified for integrity in order to ensure that the escape of biological agents and/or material into the immediate working area and outside environment is prevented. -Activities associated with the handling of live biological agents (e.g. centrifugation and blending of products which can lead to aerosol formation) should be contained -Areas where Biosafety Risk Group 3 or 4 organisms are handled should always have a negative air pressure relative to the environment. -Air-lock doors should be interlocked to avoid their being opened simultaneously. Differential pressure alarms should be present wherever required, and should be validated and monitored. -Air-vent filters should be hydrophobic and subject to integrity testing at intervals determined by QRM approach. -Where filtration of exhaust air is necessary, safe changing of filters or bag-inbag-out housings should be employed.
Next generation of flexible, modular facilities Most of vaccines today are manufactured using technologies developed 40-50 years ago. Similar antiquity, complex, uncharacterized products with high production costs. The unique nature of each vaccine manufacturing process makes it difficult to develop standard platform processes and facility designs similar to those used in Ab manufacturing Modular facilities’ design and construction with the application of single use technologies permit rapid construction and commissioning led to reducing capital and operational expenditures. Using IPV as a model From Paradigm shift for vaccine manufacturing facilities: The next generation of flexible,modular facilities: Alain Pralong et al
3.4 Use of animals(1): -Live animals should be avoided in the production area unless otherwise justified. -Embryonated eggs are allowed in production area, if applicable. -Areas used for performing tests involving animals or microorganisms, including breeding, should be well separated from premises used for manufacturing products and should have completely separate ventilation systems and separate staff. -Monitoring of compliance with TSE(transmissible spongiform encephalopathies) regulations (25), other adventitious agents that are of concern (e.g. zoonotic diseases, diseases of source animals) should also be monitored and recorded in line with specialist advice - Particular care should be taken to prevent and monitor infections in source/donor animals. - For products manufactured from transgenic animals, traceability should be maintained in the creation of such animals from the source animals. - Animals, biological agents and tests carried out should be appropriately identified to prevent any risk of mix-up and to control all identified hazards. -
Use of animals(2): - The facility layout should ensure a unidirectional and segregated flow of healthy animals, inoculated animals and waste decontamination areas. Personnel and visitors should also follow a defined flow in order to avoid crosscontamination. - For different animal species and lines, key criteria should be defined, monitored and recorded. These may include age, sex, weight and health status of the animals.
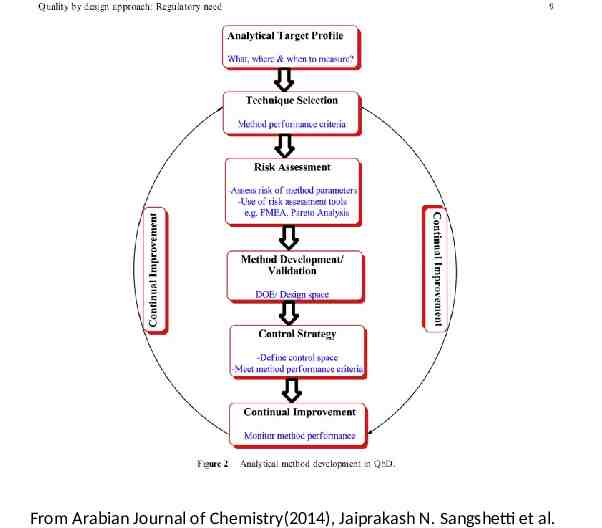
Quality by design approach “Quality could be planned and most of quality deficit arises in the way process is planned and developed” Quality Expert Joseph Moses Juran Elements of QbD: method intent, design of experiment and risk assessment More controls were required for drug manufacturing processes for efficient product and regulatory decision making Expectations were mentioned in PAT which is a system for designing, analysing, and controlling manufacturing processes based on understanding sciences and factors which affect the quality
Regulatory Aspects to QbD FDA: Q8 Pharmaceutical development Q9 Quality risk assessment Q10 Pharmaceutical Quality System QbD is a systematic approach to product and process design and development descriped in “pharmaceutical cGMPs for 21st century- a risk based approacheed
QUALITY BY DESIGN Step 1. Agree on the Target Product Profile Step 2. Determine the Critical Quality Attributes (CQAs) Step 3. Link the drug and excipient attributes and the process parameters to the CQAs Step 5. Define the Control Strategy Step 6. Prepare QbD registration file Step 7. Product lifecycle management and continual improvement EMEA/CHMP/ICH/518819/2007 Step 4. Define the Design Space
Designing and implementing control strategy Control strategy is required to ensure that material and process are within the expected lower and upper limits. Parameter and material are routinely controlled during production in order to assure reproducibility. The control space should be within the design space. Generally scale up is trial and error basis. During scale up processes parameters may differ but attributes which affect quality remain the same hence control strategy is required (Lawrence et al., 2009). QbD gives trace on reproducibility and robustness. Process capability index expresses reproducibility of process. Process capability index (CpK) (upper limit of specification - lower limit of specification)/6standard deviation
From Arabian Journal of Chemistry(2014), Jaiprakash N. Sangshetti et al.
DS Specification 1. 2. 3. 4. 5. Appearance and Description Identity(highly specific test, unique aspects) Purity and Impurities Potency Quantity
Comparability of BP subject to Changes in MP Comparing postchange to prechange product following manufacturing process changes Assessing the impact of observed differences in the quality attributes caused by the manufacturing process change for a given product as it related to safety and efficacy of the product
Summary -Evolution of BP/vaccines and production methods are intimately tied to each other -Technologies will continue to evolve as we strive for safer and more efficient products/more immunogenic vaccines with the improving and more understanding of biology -Modern vaccine development is currently exploit a wide array of novel technologies to create safer and more efficacious vaccines(viral vectors in animal cells, VLP in yeast/insect cells polysachharide conjugation to proteins, DNA plasmids in E.Coli and therapeutic cacer vaccines -Purification advances are increasing efficacy with innovative analytical methods are improving process understanding -From a regulatory perspective, Quality by Design(QbD) and Process Analytical Technology(PAT) are important initiatives that can be applied effectively to many types of vaccines and Biological Products. -CGMP regulations require that manufacturing processes be designed and controlled to assure in-process materials and finished product meet predetermined Quality requirements and do so consistently & reliably