Genomic SEM Tutorial Paper: Grotzinger, A. D., Rhemtulla, M.,












































59 Slides4.48 MB
Genomic SEM Tutorial Paper: Grotzinger, A. D., Rhemtulla, M., de Vlaming, R., Ritchie, S. J., Mallard, T. T., Hill, W. D, Ip, H. F., McIntosh, A. M., Deary, I. J., Koellinger, P. D., Harden, K. P., Nivard, M. G., & Tucker-Drob, E. M. (in press). Genomic SEM provides insights into the multivariate genetic architecture of complex traits. Nature Human Behaviour. Link to paper: rdcu.be/bvn7t
Step 0a: Load in data and package Will likely print 24 warnings about replacing previous imports: OK TO IGNORE
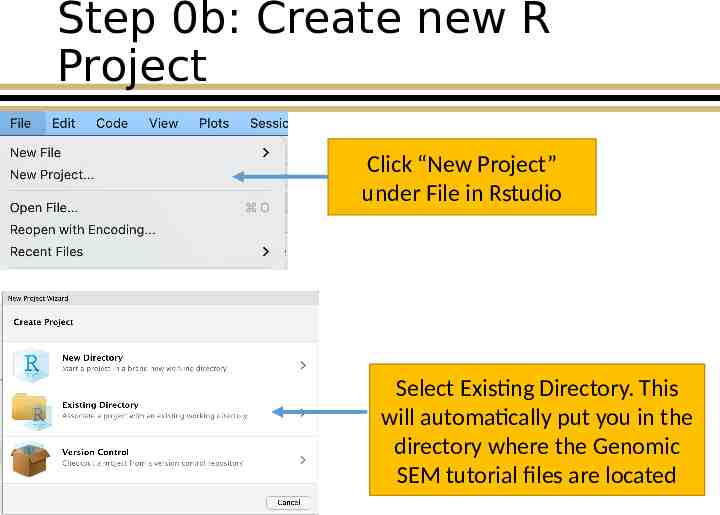
Step 0b: Create new R Project Click “New Project” under File in Rstudio Select Existing Directory. This will automatically put you in the directory where the Genomic SEM tutorial files are located
LD-score regression estimates genetic correlations between samples with varying degrees of sample overlap using publicly available GWAS data
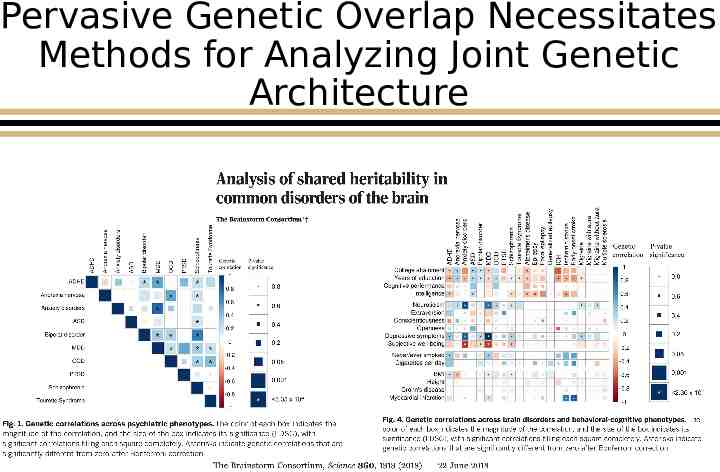
Pervasive Genetic Overlap Necessitates Methods for Analyzing Joint Genetic Architecture
Genomic SEM Genomic SEM provides flexible framework for estimating limitless number of structural equation models using multivariate genetic data from GWAS summary statistics
Practical outline I. II. III. IV. Initial considerations Estimating common factor models Estimating user specified model Estimating multivariate GWAS in Genomic SEM
I. Initial Considerations
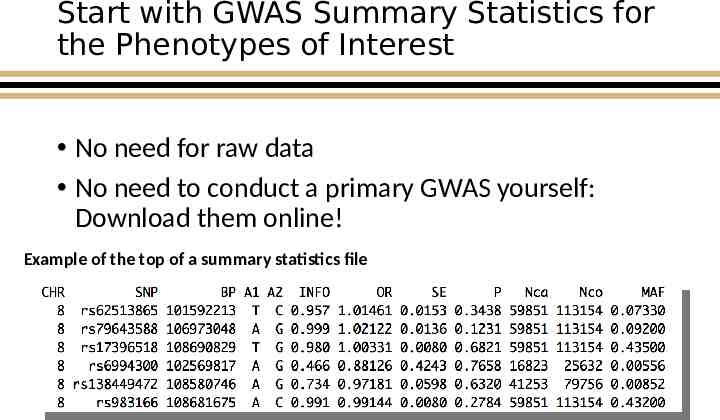
Start with GWAS Summary Statistics for the Phenotypes of Interest No need for raw data No need to conduct a primary GWAS yourself: Download them online! Example of the top of a summary statistics file
Where to get summary statistics List lots of resources on the Genomic SEM Wiki: https://github.com/GenomicSEM/GenomicSEM/wiki/2.Important-resources-and-key-information
Things to know before getting started 1. Be sure you are using summary statistics calculated within a single ethnic population Example: PTSD on PGC web-site 2. Be sure to use LD scores that match the ethnic population in sum stats 3. Typically advisable to only include summary statistics from a GWAS with N 10,000
Things to know before getting started 4. GenomicSEM allows for varying and unknown degrees of sample overlap The user does not need to know the specific levels of overlap 5. Multivariate GWAS in Genomic SEM uses listwise deletion If certain summary statistics have low genomic coverage this will affect the number of SNPs available for all included traits 6. Make sure you are not using a pruned list of summary statistics (e.g., the top 5,000 hits)
Things to know before getting started 7. Both the munge and sumstats functions in GenomicSEM use sample size to perform necessary conversions. Sample size from summary statistics file or provided by the user. In order to produce accurate results, this should be the total sample size for all included traits. Be wary of: Publicly available summary statistics that exclude certain cohorts (e.g., 23andMe).
Questions from videos?
II. Estimating Common Factor Models in Genomic SEM
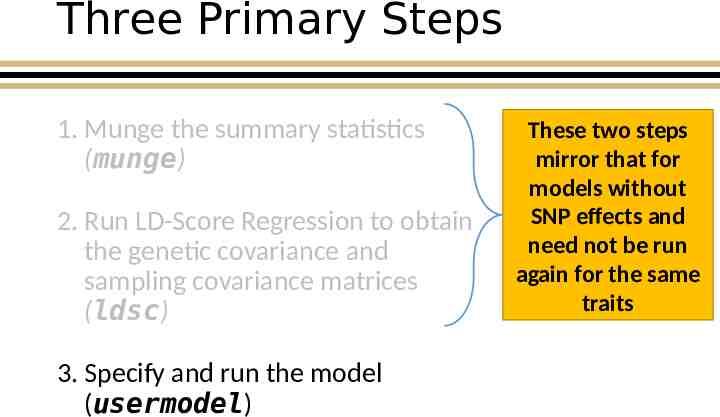
Three Primary Steps 1. Munge the summary statistics (munge) 2. Run LD-Score Regression to obtain the genetic covariance and sampling covariance matrices (ldsc) 3. Run the model (commonfactor) Munge: convert raw data from one form to another
Lab Using GWAS sumstats for: Schizophrenia (Pardiñas et al., 2018); N 105,318 Bipolar Disorder (Sklar et al., 2011); N 16,731 Major Depressive Disorder (Wray et al., 2018); N 173,005
Step 1: munge example code Note that for much of the practical we are using a subset of the summary statistics for demonstrative purposes only! In practice, you will use the full set of summary stats you download online
Examine SCZ BIP MDD munge.log file
Step 2: ldsc example code Populated with ld scores from the same ancestry
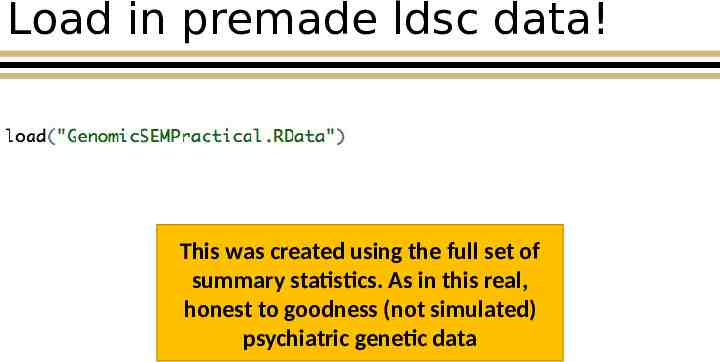
Load in premade ldsc data! This was created using the full set of summary statistics. As in this real, honest to goodness (not simulated) psychiatric genetic data
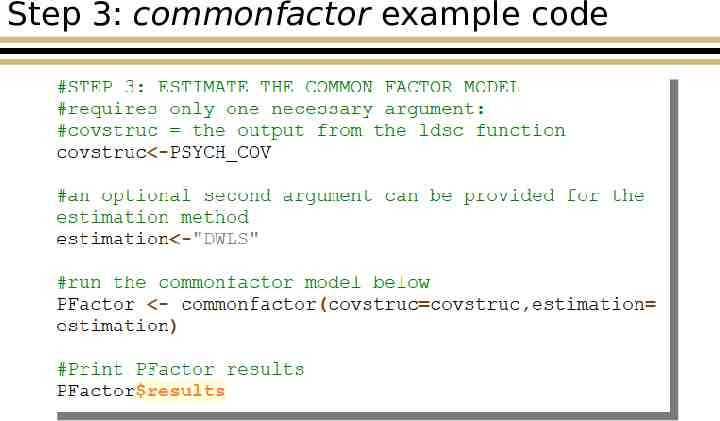
Step 3: commonfactor example code
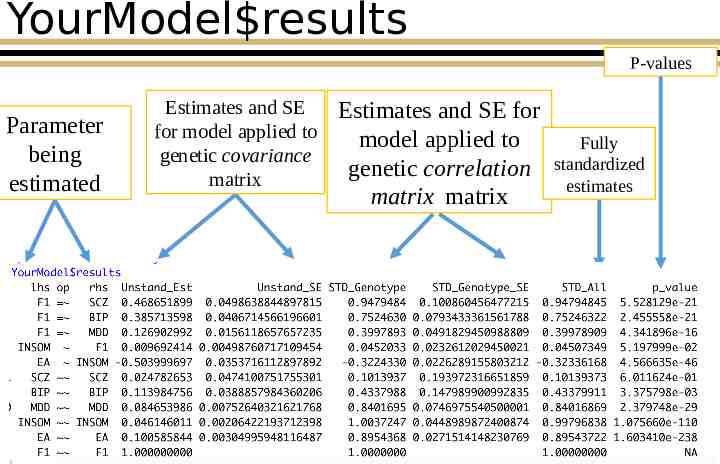
Pfactor results Parameter being estimated Estimates and SE for model applied to genetic covariance matrix Estimates and SE for model applied to genetic correlation matrix
Go to breakout rooms
III. Estimate a UserSpecified Model
Three Primary Steps 1. Munge the summary statistics (munge) 2. Run LD-Score Regression to obtain the genetic covariance and sampling covariance matrices (ldsc) 3. Specify and run the model (usermodel) These two steps mirror that for models without SNP effects and need not be run again for the same traits
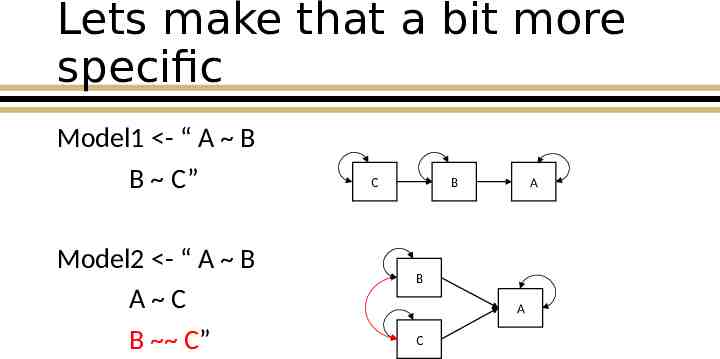
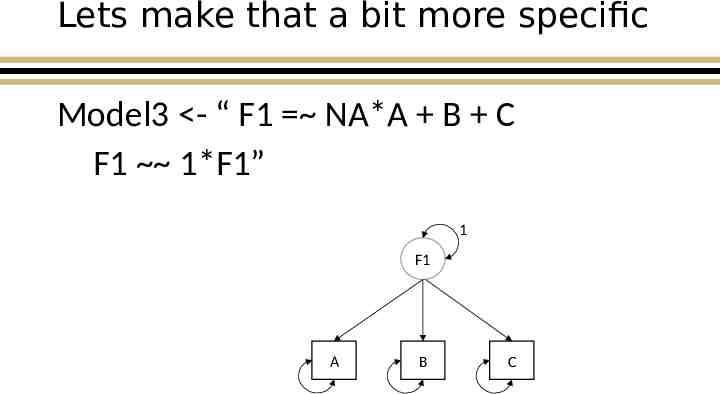
How to specify a model We use the lavaan formula language, slightly extended: Regression: A B (Co)variance: A A; A B Factor: F1 A B C D Fix a parameter: A 1*B (the covariance between A and B is 1) Name a parameter: A a*B (the covariance between A and B parameter label a) Allows you to use model constraints for this parameter: a .001
Lets make that a bit more specific Model1 - “ A B B C” Model2 - “ A B A C B C” C B A B A C
Lets make that a bit more specific Model3 - “ F1 NA*A B C F1 1*F1” 1 F1 A B C
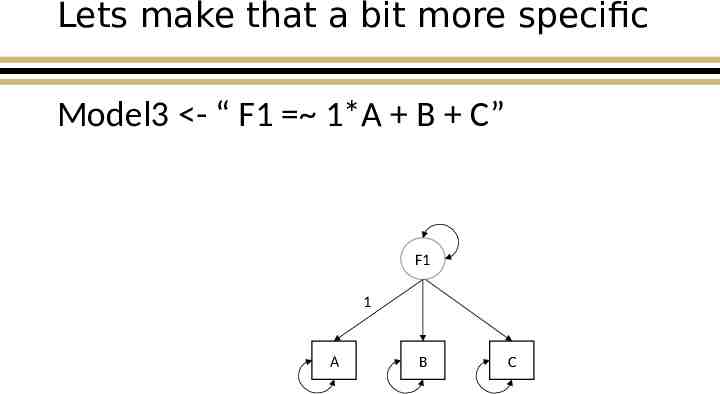
Lets make that a bit more specific Model3 - “ F1 1*A B C” F1 1 A B C
Lab Used GWAS sumstats for: Schizophrenia (Pardiñas et al., 2018); N 105,318 Bipolar Disorder (Sklar et al., 2011); N 16,731 Major Depressive Disorder (Wray et al., 2018); N 173,005 Educational Attainment (Lee et al., 2019); N 766,035 Insomnia (Jansen et al., 2019); N 386,533
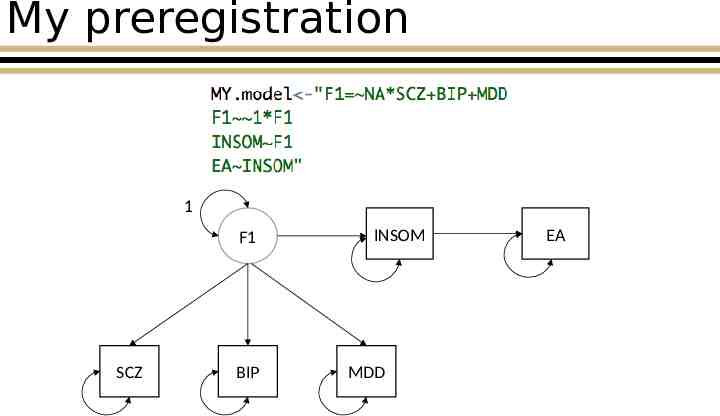
My preregistration 1 F1 SCZ BIP INSOM MDD EA
Specify Arguments
YourModel results P-values Parameter being estimated Estimates and SE for model applied to genetic covariance matrix Estimates and SE for model applied to genetic correlation matrix matrix Fully standardized estimates
YourModel modelfit chisq: The model chi-square, reflecting index of exact fit to observed data, with lower values indicating better fit. df and p chisq: The degrees of freedom and p-value for the model chi-square. AIC: Akaike Information Criterion. Can be used to compare models regardless of whether they are nested. CFI: Comparative Fit Index. Higher better. .90 acceptable fit; .95 good model fit SRMR: Standardized Room Mean Square Residual. Lower better. .10 acceptable fit; .05 good fit
Delete Input for MY.model and run your own!
PRACTICAL: You Take Control As away of preregistering them, discuss the model as a group and select someone to report the model you ran after the breakout room Remember five variable names are: SCZ, BIP, MDD, EA, INSOM Also, again, this is real data. Your discoveries are REAL!
Go to breakout rooms
Models run by different breakout rooms?
IV. Multivariate GWAS in Genomic SEM
Four Primary Steps 1. Munge the summary statistics (munge) 2. Run LD-Score Regression to obtain the genetic covariance and sampling covariance matrices (ldsc) 3. Prepare the summary statistics for multivariate GWAS (sumstats) 4. Run the multivariate GWAS (commonfactorGWAS; userGWAS) These two steps mirror that for models without SNP effects and need not be run again for the same traits
Lab Using Same GWAS sumstats for: Schizophrenia (Pardiñas et al., 2018); N 105,318 Bipolar Disorder (Sklar et al., 2011); N 16,731 Major Depressive Disorder (Wray et al., 2018); N 173,005
Step 3: sumstats example code Flowchart on github to help you figure out arguments for sumstats
Examine SCZ BIP MDD sumstats.log file
Behind the scenes GenomicSEM GWAS functions automatically combine output from Steps 2 and 3 Creates as many covariance matrices as there are SNPs across traits Step 3: Run sumstats GWAS functions combine the two Step 2: Run ldsc
Step 4a: commonfactorGWAS example code To save memory, saves only the effect of the SNP on the common factor
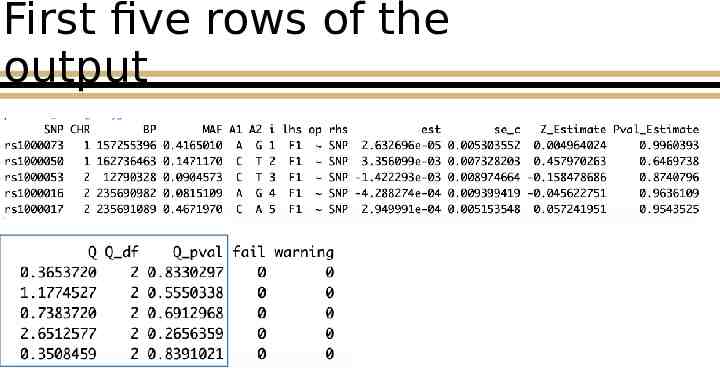
First five rows of the output
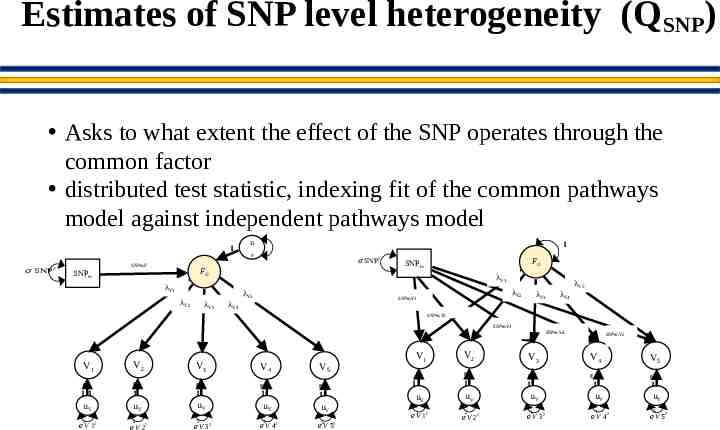
Estimates of SNP level heterogeneity (QSNP) Asks to what extent the effect of the SNP operates through the common factor distributed test statistic, indexing fit of the common pathways model against independent pathways model 1 𝜎 SNP 2 SNPm,F u 1 F 𝜎 SNP FG SNPm λV5 λV3 FG SNPm λV1 λV1 λV2 2 λV4 λV5 λV2 SNPm,V1 λV3 λV4 SNPm,V2 SNPm,V3 SNPm,V4 V1 V2 V3 V4 V5 g g g g g 11 1 1 1 1 uV uV uV 1 2 𝑒 V 12 𝑒V 2 2 3 𝑒 V 32 uV 4 𝑒V 42 uV 𝑒5 V 52 V1 V2 g 1 uV 1 𝑒 V 12 SNPm,V5 V3 V4 V5 g g g g 1 1 uV 2 𝑒 V 22 uV 3 𝑒V 3 2 1 1 uV uV 4 𝑒 V 42 5 𝑒V 52
Troubleshooting
Step 4b: userGWAS example code
Step 4b: userGWAS example code
If you finish early feel free to go onto next slides/section of code to play around more with specifying your own model
Go to breakout rooms
If there’s time play around with some anthropometric traits Note that you do not need to include all variables in the model
Variable Names BMI Body Mass Index WHR Waist Hip Ratio Waist Waist Circumference Hip Hip circumference CO childhood obesity Height Height BL Birth Length BW Birth Weight IHC Infant Head Circumference
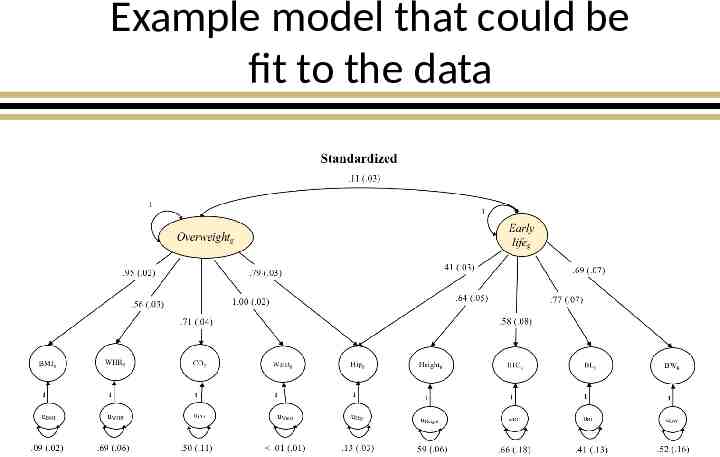
Example model that could be fit to the data
Final Notes Parallel processing for both userGWAS and commonfactorGWAS is available Parallel is the same as serial processing, except that it takes an additional cores argument specifying how many cores to use Ideal run-time scenario: split jobs across computing nodes on a cluster and run in-parallel All runs are independent of one another!
Overview Genomic SEM is ready for use today! Ask questions on our google forum https://groups.google.com/forum/#!forum/genomic-sem-users Lots can be done using existing, openly available GWAS summary statistics Models are flexible and up to the user Use Genomic SEM to derive sumstats for novel phenotypes for use in PGS analyses
Resources See paper at: rdcu.be/bvn7t See github at: https://github.com/GenomicSEM/GenomicSEM See tutorials at: https://github.com/GenomicSEM/GenomicSEM/wi ki






